Events
Talks, exhibitions and happenings-
 AGE OF WONDER 2014
AGE OF WONDER 2014 -
 ART AND INNOVATION: THE FUTURE OF INTERDISCIPLINARITY 2014
ART AND INNOVATION: THE FUTURE OF INTERDISCIPLINARITY 2014 -
 LIVING MIRROR 2013
LIVING MIRROR 2013 -
 TERRA 0 2013
TERRA 0 2013 -
 GROW YOUR OWN 2013
GROW YOUR OWN 2013 -
 THE FUTURE OF ART-SCIENCE COLLABORATIONS 2013
THE FUTURE OF ART-SCIENCE COLLABORATIONS 2013 -
 AMOLF OPEN DAY 2013
AMOLF OPEN DAY 2013 -
 NOT INVENTED BY NATURE: HELMHOLTZ INITIATIVE SYNTHETIC BIOLOGY 2013
NOT INVENTED BY NATURE: HELMHOLTZ INITIATIVE SYNTHETIC BIOLOGY 2013 -
 DA4GA OPENING EXHIBITION 2013
DA4GA OPENING EXHIBITION 2013 -
 ARTSCIENCE TALKSHOW 2013
ARTSCIENCE TALKSHOW 2013 -
 ART-SCIENCE TALK 2013
ART-SCIENCE TALK 2013 -
 SPEED DEBATE ON SYNTHETIC BIOLOGY AND NEURO-ETHICS 2013
SPEED DEBATE ON SYNTHETIC BIOLOGY AND NEURO-ETHICS 2013 -
 YOUNG SYNTHETIC BIOLOGISTS - YSB 1.0 2013
YOUNG SYNTHETIC BIOLOGISTS - YSB 1.0 2013 -
 EN VIE / ALIVE 2013
EN VIE / ALIVE 2013 -
 DO IT TOGETHER BIO #5: SYNTHETIC BIOLOGY ART 2013
DO IT TOGETHER BIO #5: SYNTHETIC BIOLOGY ART 2013 -
 ART FROM SYNTHETIC BIOLOGY 2013
ART FROM SYNTHETIC BIOLOGY 2013 -
 EVAPORATION OF THINGS 2013
EVAPORATION OF THINGS 2013 -
 DESIGNERS & ARTISTS 4 GENOMICS AWARD CEREMONY 2012
DESIGNERS & ARTISTS 4 GENOMICS AWARD CEREMONY 2012 -
 MUTAMORPHOSIS: TRIBUTE TO UNCERTAINTY 2012
MUTAMORPHOSIS: TRIBUTE TO UNCERTAINTY 2012 -
 SYNTHETIC BIOLOGY SOCIETY KICK OFF EVENT 2012
SYNTHETIC BIOLOGY SOCIETY KICK OFF EVENT 2012 -
 RE-NEW DIGITAL ARTS FESTIVAL 2012
RE-NEW DIGITAL ARTS FESTIVAL 2012 -
 CAGE RATTLING #1: KILL SWITCH 2012
CAGE RATTLING #1: KILL SWITCH 2012 -
 RIGHT OR RISK? WORLD'S FIRST PUBLIC BIOBRICK 2012
RIGHT OR RISK? WORLD'S FIRST PUBLIC BIOBRICK 2012 -
 SYNTHETIC BIOLOGY SPEED DEBATE 2012
SYNTHETIC BIOLOGY SPEED DEBATE 2012 -
 GRADUATE SCHOOL LAUNCH 2012
GRADUATE SCHOOL LAUNCH 2012 -
 ARTISTS IN LABORATORIES 2012
ARTISTS IN LABORATORIES 2012 -
 SUBTLE TECHNOLOGIES 2012
SUBTLE TECHNOLOGIES 2012 -
 THE TWO CULTURES: VISUAL ART AND THE SCIENCES C.1800-2011 2012
THE TWO CULTURES: VISUAL ART AND THE SCIENCES C.1800-2011 2012 -
 C-LAB PREMIERS LIVING SYNTHETIC BIOLOGY WORKS AT TECHFEST 2012 2012
C-LAB PREMIERS LIVING SYNTHETIC BIOLOGY WORKS AT TECHFEST 2012 2012 -
 ANTÓNIO CARAMELO'S DREAMING OF A BUTTERFLY 2011
ANTÓNIO CARAMELO'S DREAMING OF A BUTTERFLY 2011 -
 SYNTHETIC BIOLOGY: MACHINE OR LIFE? 2011
SYNTHETIC BIOLOGY: MACHINE OR LIFE? 2011 -
 ANNE BRODIE'S BEE BOX 2011
ANNE BRODIE'S BEE BOX 2011 -
 EUROPEAN PUBLIC ART CENTRE: EPAC 2011
EUROPEAN PUBLIC ART CENTRE: EPAC 2011 -
 SYNTHESIS: SYNTHETIC BIOLOGY IN ART & SOCIETY 2011
SYNTHESIS: SYNTHETIC BIOLOGY IN ART & SOCIETY 2011 -
 CABI OFFICIAL OPENING 2011
CABI OFFICIAL OPENING 2011 -
 VIVA EXPERIHIBITION 2011
VIVA EXPERIHIBITION 2011 -
 DIGITAL MATTER RESEARCH FORUM 2010
DIGITAL MATTER RESEARCH FORUM 2010 -
 BIOART FORUM 2010
BIOART FORUM 2010 -
 INTO THE LABS 2009
INTO THE LABS 2009 -
 (UN)INHABITABLE? – ART OF EXTREME ENVIRONMENTS 2009
(UN)INHABITABLE? – ART OF EXTREME ENVIRONMENTS 2009 -
 THE MARS SIMULATION LABORATORY 2009
THE MARS SIMULATION LABORATORY 2009 -
 TRANSIENT CREATURES 2008
TRANSIENT CREATURES 2008 -
 LESS REMOTE: THE FUTURES OF SPACE EXPLORATION 2008
LESS REMOTE: THE FUTURES OF SPACE EXPLORATION 2008 -
 TELLING PLACES: NARRATIVE AND IDENTITY IN ART AND ARCHITECTURE 2007
TELLING PLACES: NARRATIVE AND IDENTITY IN ART AND ARCHITECTURE 2007 -
 UNSAFE DISTANCE 2007
UNSAFE DISTANCE 2007 -
 MUTAMORPHOSIS: CHALLENGING ARTS AND SCIENCES 2007
MUTAMORPHOSIS: CHALLENGING ARTS AND SCIENCES 2007 -
 ROCHE CONTINENTS 2007
ROCHE CONTINENTS 2007 -
 BIORAMA SESSIONS 2007
BIORAMA SESSIONS 2007 -
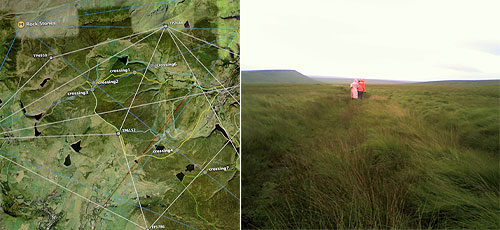 BIORAMA HIKE 2007
BIORAMA HIKE 2007 -
 BIOS 4. ARTE BIOTECNOLÓGICO Y AMBIENTAL 2007
BIOS 4. ARTE BIOTECNOLÓGICO Y AMBIENTAL 2007 -
 AN ENCOUNTER WITH BIOTECHNOLOGICAL AND ENVIRONMENTAL ART 2007
AN ENCOUNTER WITH BIOTECHNOLOGICAL AND ENVIRONMENTAL ART 2007 -
 EXPOSING ROSES TO MARTIAN CONDITIONS AT THE MARS LAB 2007
EXPOSING ROSES TO MARTIAN CONDITIONS AT THE MARS LAB 2007 -
 INTRODUCTION TO BACTERIOLOGICAL METHODS 2006
INTRODUCTION TO BACTERIOLOGICAL METHODS 2006 -
 FLESHING OUT 2006
FLESHING OUT 2006 -
 C-LAB VISITS CALAR ALTO 2006
C-LAB VISITS CALAR ALTO 2006 -
 GLASS BODY 2006
GLASS BODY 2006 -
 BIOART :: LECTURE 2006
BIOART :: LECTURE 2006 -
 C-LAB VISITS THE MARSLAB 2005
C-LAB VISITS THE MARSLAB 2005 -
 OFFICIAL LAUNCH OF THE ARTS AND GENOMICS CENTRE 2005
OFFICIAL LAUNCH OF THE ARTS AND GENOMICS CENTRE 2005 -
 TRANSGENIC ART FORUM 2005
TRANSGENIC ART FORUM 2005 -
 BIOTECH ART WORKSHOP 2005
BIOTECH ART WORKSHOP 2005 -
 TODAY IN PARADISE - GENETICS & ARTS 2005
TODAY IN PARADISE - GENETICS & ARTS 2005 -
 SYMPOSIUM 2005
SYMPOSIUM 2005 -
 THE MEXICO PROJECT :: TALK 2004
THE MEXICO PROJECT :: TALK 2004 -
 C-LAB VISITS MIT 2004
C-LAB VISITS MIT 2004 -
 THE CACTUS PROJECT :: LECTURE 2002
THE CACTUS PROJECT :: LECTURE 2002